10.1 Parte 1: Análisis univariados a datos de estructura multivariada
Se requiere identificar si existen cambios en la composición de especies bentónicas en una línea de costa en cuyo extremo oriental se están vertiendo aguas residuales de una planta de tratamiento de un gran hotel. Estas aguas, aun siendo tratadas, siguen siendo ricas en materia orgánica, y se dispersan según el patrón de corrientes en sentido Este-Oeste. En principio, la línea de costa es bastante homogénea en características ambientales, siendo la contaminación orgánica la única variable que genera gradiente ambiental. Se presume que el aporte orgánico puede estar modificando la estructura trófica del ecosistema marino-costero según el gradiente de dispersión. Para identificar si esto ocurre, se decidió evaluar la composición de especies y las abundancias de los poliquetos de la macrofauna, ya que es bien conocido que son excelentes bioindicadores de contaminación orgánica. Se tomaron 10 muestras de sedimento en seis localidades a lo largo de la bahía, desde el punto de vertido hasta 30 km alejados de la fuente de contaminación. Las abundancias por especies fueron agrupadas por localidad y se encuentran en el archivo “gradiente.csv”. La actividad consiste en identificar el gradiente biológico. NOTA: Sí existe gradiente biológico, fallar en detectarlo implica librar al hotel de responsabilidades ambientales y administrativas ante el gobierno.
Importe la matriz de datos “gradiente.csv” y nómbrela gradiente. Examine el objeto y responda:
Region Localidades sp1 sp2 sp3 sp4 sp5 sp6 sp7 sp8 sp9 sp10
1 1 Loc1 16 NA NA 3 210 31 NA 5 NA NA
2 1 Loc2 32 NA NA 4 240 NA NA 7 NA 22
3 2 Loc3 NA 13 1 NA 199 24 NA 1 NA NA
4 2 Loc4 1 6 NA NA 200 23 2 NA NA NA
5 3 Loc5 NA 31 1 NA 173 NA 3 NA 12 NA
6 3 Loc6 NA 15 3 NA 205 NA 4 NA 9 NA
¿Qué tipo de objeto se creó? Si usaste la función read_csv del paquete readr, asegura de transformar el objeto resultante a data frame con la función as.data.frame.
¿Cuáles son las dimensiones del objeto?
¿qué representan las filas y qué representan las columnas?
¿Con base en las dimensiones, se puede inferir cuántas especies hay en la matriz y en cada localidad?
¿Son similares las magnitudes de abundancia de las especies en cada localidad?
Considerando que esta es una matriz pequeña ¿puede apreciar algún patrón biológico viendo solo la tabla?
Genere una tabla de nombre “uni” que describa las propiedades UNIVARIADAS bióticas de las localidades, incluya: la riqueza de especies, la abundancia de individuos y el índice de diversidad de Simpson. Le recomiendo buscar ayuda sobre las funciones specnumber, diversity. Acá un script que le ayudará a avanzar rápido. Nota: ninguna de estas funciones filtran texto
¿Obtuvo un error? ¿qué cree que pudo ocurrir?
Seleccione sólo las columnas con información de las abundancias de las especies y llámelas “dat”.
Repita el script para generar la tabla de descriptores univariados.
Calculados los descriptores univariados ¿puede apreciar algún patrón biológico viendo solo la tabla? Explique el patrón. Si considera necesario realizar algún gráfico, hágalo. El siguiente script puede ser útil, pero no es la única forma.
Aparentemente no hay un patrón biológico según el gradiente, sin embargo, la ausencia de patrón puede ser producto del reduccionismo de los descriptores univariados. Por ello, usaremos métodos multivariados para apreciar mejor la informacion de la matriz.
10.2 Parte 2: Análisis en modo Q
Hemos identificado que nuestra matriz tiene 6 muestras (localidades) y 10 especies. Además, no todas las especies están en todas las muestras, hay especies con abundancias muy elevadas y otras muy bajas. ¿qué representa esto para iniciar un análisis multivariado? Exploremos esto usando varios métodos. Sugiero leer el material de ayuda vegdist del paquete vegan, tipeando ??vegdist en la cónsola. Para esta práctica compararemos el desempeño del índice Bray-Curtis y la distancia Euclideana.
Iniciemos con las distancias Euclideanas. Use la función vegdist, y llame al resultado euc.
¿cuántos valores se estimaron teniendo en cuenta que hay 6 localidades?
¿Qué tipo de objeto es bray1?
¿qué fue lo que se estimaron, similitudes o dismilitudes? ¿cómo se interpretan estas unidades? ¿cuál es su escala? Para obtener las similitudes e interpretarlas con mayor facilidad aplique el siguiente comando
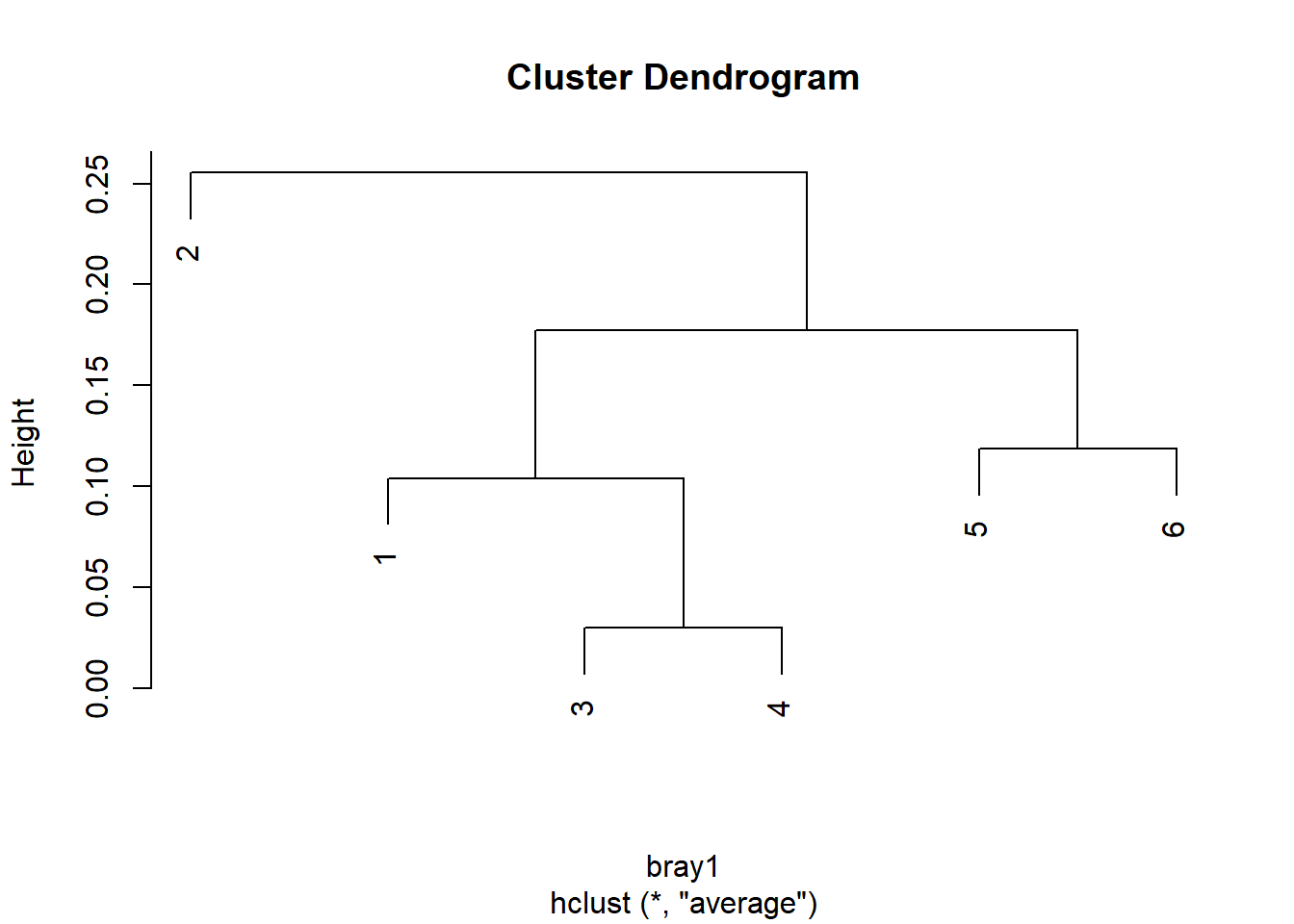
¿cuáles son las localidades más parecidas? ¿qué puede decir de la similitud entre la localidad 1 y 6? ñ) ¿Aprecia el gradiente? Si no, genere un dendograma que nos ayude a visualizar el patrón de cambio
Código
#|include: falseplot(hclust(bray1, "average"))
Este gráfico le permitirá identificar el par de localidades, o subgrupos de localidades, más semejantes en forma jerárquica y pareada ¿Aprecia el patrón?
El patrón aun no se aprecia, probablemente se deba al peso de la especie 5. Aplique transformación raíz cuadrada a las abundancias y llame a esa matriz nueva “dat2”. Calcúle nuevamente el índice Bray-Curtis y llámelo bray2.
¿cuáles con las localidades más parecidas? ¿qué puede decir de la similitud entre la localidad 1 y 6? ¿Aprecia el gradiente? Genere un dendograma que nos ayude a visualizar el patrón de cambio
Código
plot(hclust(bray2, "average"))
¿Ahora sí aprecia el patrón de cambio con el gradiente ambiental?
Compare los tres dendogramas
Genere un nuevo gráfico multivariado, es un nMDS (escalamiento multidimensional no métrico - tema de la Unidad 5). Por ahora solo importa indicar que la distancia de los puntos proyectados en el nMDS refleja las similitudes expresadas en la matriz. En este sentido ¿aprecia mejor el gradiente de cambio en la composición de especies?
Código
mds <-metaMDS(dat2, "bray")plot(mds, type ="n")text(mds,display ="sites",cex =1,col ="blue")
El enunciado hace referencia a la composición de especies, explícitamente indica que solo importa el cambio en la identidad de las especies, no en sus abundancias. Esto implica que a pesar de la transformación raíz cuadrada, las diferencias en abundancias pueden estar influyendo en el patrón observado. Para satisfacer plenamente la pregunta enunciada, se aplicará un índice que sólo considere la presencia de la especie. Hay más de 25 índices para evaluar matrices de incidencia, uno de los más populares es Jaccard. Aplique este índice con la función vegdist y llame al objeto “jac”.
¿Qué puede inferir ahora del patrón y el gradiente? ¿cuál es la similitud en composición de especies entre las localidades 1 y 6?
¿Cuál es la similtud en la composición de especies entre las localidades 6 y 5?
Genere un dendograma para proyectar la nueva matriz. Interprete el agrupamiento del dendograma
Veamos ahora globalmente los cuatro dendogramas generados. Para ello use el siguiente código
Código
par(mfrow =c(2, 2), mar =c(2, 4, 2, 2)) # este script indica a R que se#proyectarán cuatro gráficos en uno soloplot(hclust(euc, "average"), main ="Dist. Euclideana")plot(hclust(bray1, "average"), main ="Bray-curtis sin transformación")plot(hclust(bray2, "average"), main ="Bray-curtis con transformación")plot(hclust(jac, "average"), main ="Jaccard")
Existes métodos cuantitativos para comparar matrices. Uno de ellos es el método de Mantel, y se base en correlacionar los rangos de las jerarquias de las similitudes en cada matríz. Por ahora solo interesa conocer la correlación, y no la probabilidad asociada. Como reto para usted, busque en el paquete vegan la función mantel, y correlacione la matríz de similitud en composición de especies respecto a las matrices Bray-Curtis y la Matriz de distancias Eucliedanas.